ANNOVAR是华人王凯写的用于snp等变异位点注释的软件,说实话,这个东东还真难下载,摸索了好几天,总算中了彩票。
ANNOVAR的下载
下载地址在:
http://annovar.openbioinformatics.org/en/latest/user-guide/download/
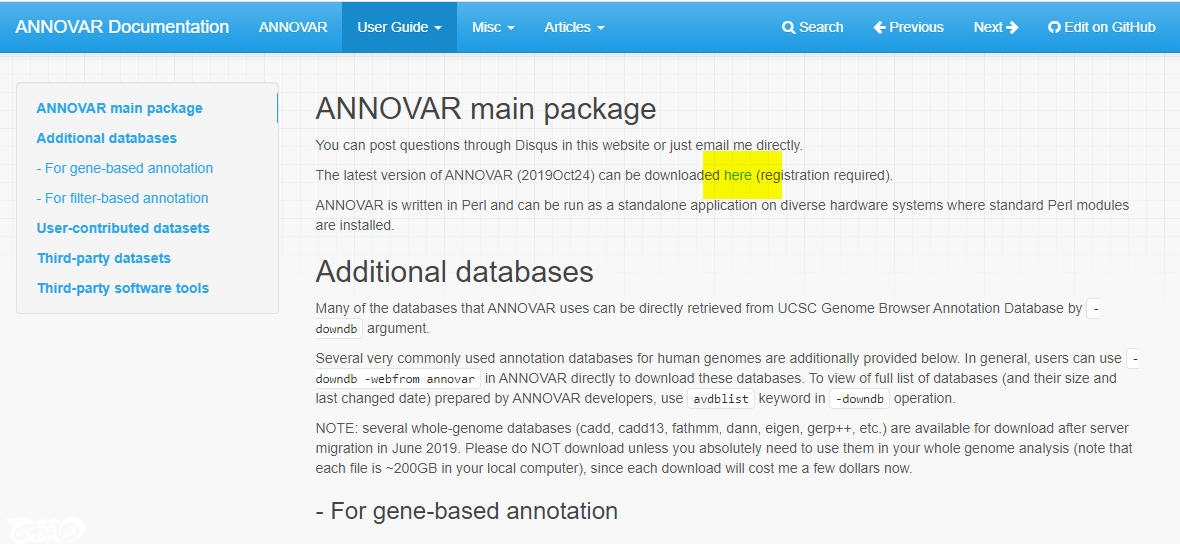
点击图中的“here”打开下面的这个提交地址
提交邮箱及其它信息地址:
http://download.openbioinformatics.org/annovar_download_form.php
ANNOVAR 的文件结构
ANNOVAR │ annotate_variation.pl #主程序,功能包括下载数据库,三种不同的注释 │ coding_change.pl #可用来推断蛋白质序列 │ convert2annovar.pl #将多种格式转为.avinput的程序 │ retrieve_seq_from_fasta.pl #用于自行建立其他物种的转录本 │ table_annovar.pl #注释程序,可一次性完成三种类型的注释 │ variants_reduction.pl #可用来更灵活地定制过滤注释流程 ├─example #存放示例文件 └─humandb #人类注释数据库
对于非人类物种注释,最重要的就是convert2annovar.pl、retrieve_seq_from_fasta.pl、annotate_variation.pl和table_annovar.pl这四个核心脚本了。
两个关键点:
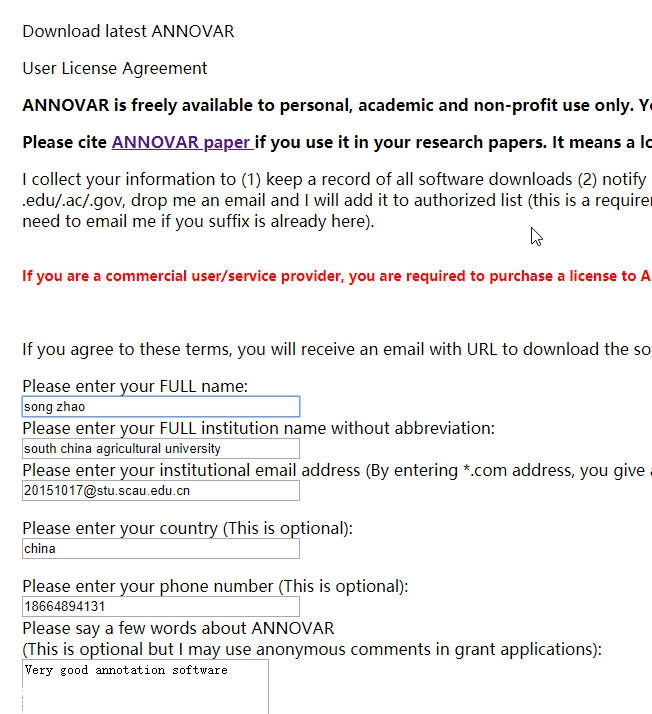
1,这里的email需要填写edu.cn结尾的邮箱,
2,openbioinformatics.org的这两个地址需要翻墙才能打开。
所以这个问题是导致我找了老半天才下载到这个软件的原因,开始还以为那个地址作废了,一次一次重复去找,真没想到是firewall的原因。
既然这样,那就把我下载的annovar.latest分享给大家,从文件日期来看,这个是2020年6月8号修改的最新版。
删除了humandb文件夹中的数据库哈,因为我主要分析辣椒的。
在conda中有一个jannovar库,也可以用来注释vcf
conda install jannovar-cli
添加环境变量
annovar的脚本均为perl编写,和sh脚本一样,那么是不能直接运行这个pl脚本的,都需要在脚本的前面加上perl字样,代表这是一个pl脚本,比如retrieve_seq_from_fasta.pl,在运行的时候需要添加perl
perl retrieve_seq_from_fasta.pl --format refGene --seqfile zunla.fasta zunla_refGene.txt --out zunla_refGeneMrna.fa
但是这对于操作来说不方便,我们可以把这个pl脚本的目录添加到系统的环境变量$path中,这样以后就可以直接执行这个目录的pl脚本了,而不需要添加perl字样。具体怎么做呢?修改bashrc文件。
比如我的annovar的路径是/www/annovar
我是用root登录的,那么就直接修改root的bashrc文件
输入命令
vi ~/.bashrc
或者
vi /root/.bashrc
都是一样。
之后在文件的最下面插入代码
export PATH=”/www/annovar:$PATH”
保存,退出
执行命令
source ~/.bashrc
重载当前配置,马上生效!
构建自定义注释库
ANNOVAR可以从服务器下载注释库,但是主要针对人类基因组,那么需要分析、注释其它的物种测序结果,怎么办呢,需要自建注释库。
首先需要参考基因组的序列和注释文件,我这里是名为zunla辣椒
zunla.fasta #zunla的参考基因组序列 zunla.gff3 #zunla的注释文件,我这里是gff3
ANNOVAR建库需要genePred文件,因而需要准备几个转换gff3到genePred格式的软件
gffread #gff3 to gtf gtfToGenePred #gtf to genePred
gffread的下载地址:https://github.com/gpertea/gffread
需要自行编译
git clone https://github.com/gpertea/gffread cd gffread make release
编译过程会自行下载https://github.com/gpertea/gclib,也可以预先下载,然后make。
这里推荐直接conda安装
conda install gffread
gtfToGenePred的下载地址:
http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/gtfToGenePred
已经编译好了,下载直接使用,他属于UCSC工具包,因而在conda环境安装gtfToGenePred的命令为
conda install ucsc-gtftogenepred
conda直接安装gtftogenepred没成功,需要加上UCSC前缀。
在ANNOVAR文件夹中建立辣椒注释库的文件夹pepperdb,然后进去
mkdir pepperdb
cd pepperdb
第一步:转换gff3 2 gtf
gffread zunla.gff -T -o zunla.gtf
第二步:转换gtf 2 GenePred
gtfToGenePred -genePredExt zunla.gtf zunla.txt
第三步:建立注释库
运行命令
perl ../retrieve_seq_from_fasta.pl --format refGene --seqfile zunla.fasta zunla_refGene.txt --out zunla_refGeneMrna.fa
这样我们的建库就完成了,获得两个重要的文件
zunla_refGeneMrna.fa zunla_refGene.txt
强调一点:
关于文件的命名,看了好几篇文献,都没有说这个注释库两个文件命名的事情,按照常规逻辑,这两个文件肯定不能随便命名,不然annovar无法识别!摸索了一下,前缀就是下面build参数使用的名字,这里就是zunla,下面注释就要使用“-build zunla”这个参数,对于基于基因注释的txt文件命名就是refGene,连起来就是 zunla_refGene.txt。而fa文件前缀一样,后面有稍稍差别为refGeneMrna,连起来就是zunla_refGeneMrna.fa。
转换需要注释的vcf文件
BB3.HC.vcf是HaplotypeCaller得到的vcf文件,转换成适用annovar的文件格式
执行命令
perl ../convert2annovar.pl -format vcf4 BB3.HC.vcf > BB3.avinput
那么BB3.avinput就是我们需要注释的文件
annotate_variation注释
来到annovar的根目录,用annotate_variation.pl进行基于基因(gene-based annotation)的注释,
perl annotate_variation.pl -geneanno -dbtype refGene -out BB3 -build zunla BB3.avinput pepperdb/ # -geneanno 表示使用基于基因的注释 # -dbtype refGene 表示使用"refGene"类型的数据库 # -out zunla 表示输出以BB3为前缀的结果文件
-geneanno -dbtype refGene是默认值,可以省略,那么命令也可以写成
perl annotate_variation.pl -out BB3 -build zunla BB3.avinput pepperdb/
得到结果文件:
BB3.exonic_variant_function #外显子区域突变的功能、类型等 BB3.variant_function#突变的基因及位置 BB3.log#日志文件
table_annovar.pl注释
table_annovar.pl是ANNOVAR多个脚本的封装,可以一次性完成三种类型的注释。
我这里只有regGene类型的注释库,那么注释命令为
perl table_annovar.pl BB3.avinput pepperdb/ -buildver zunla -out BB3 -remove -protocol refGene -operation g -nastring . -csvout
-csvout #输出csv文件
-protocol refGene #注释库类型为refgene
-operation g #操作子为g
-nastring . #缺省值用“.”代替
-buildver zunla #使用zunla注释库
-out BB3 #输出文件名以BB3开头
-remove #清除所有临时文件
table_annovar.pl也可以不经过转换,直接对vcf文件进行注释,添加-vcfinput参数就行,但是需要注意的是不能和-csvout参数同时使用,命令如下
perl table_annovar.pl BB3.HC.vcf pepperdb/ -buildver zunla -out BB3 -remove -protocol refGene -operation g -nastring . -vcfinput -polish
执行命令时是出现“权限不够”错误
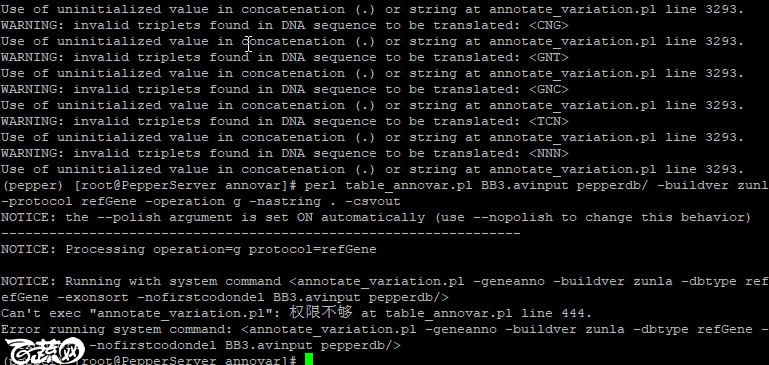
执行命令:
chmod 777 table_annovar.pl
问题依旧!
没办法,只能跑去table_annovar.pl脚本的444行看一下具体代码
#generate gene anno
my $sc;
$sc = $SC_PREFIX . "annotate_variation.pl -geneanno -buildver $buildver -dbtype $protocol -outfile $tempfile.$protocol -exonsort -nofirstcodondel $queryfile $dbloc"; #20191010: add -nofirstcodondel
$argument and $sc .= " $argument";
$intronhgvs and $sc .= " -splicing_threshold $intronhgvs";
if ($thread) {
$sc .= " -thread $thread";
}
if ($maxgenethread) {
$sc .= " -maxgenethread $maxgenethread";
}
print STDERR "\nNOTICE: Running with system command <$sc>\n";
system ($sc) and die "Error running system command: <$sc>\n";
444行的内容为system ($sc) and die "Error running system command: <$sc>\n";
看得出来,这里执行了system系统命令,往上查找变量$sc的值,发现这里调用了annotate_variation.pl脚本,同样
chmod 777 annotate_variation.pl
为了避免别的脚本权限问题,干脆全部777
chmod 777 *
成功解决权限问题。
转载请注明:百蔬君 » 【原创文章】用ANNOVAR自建数据库注释辣椒高通量序列